Our group uses computer modelling tools to study biophysical chemistry. We integrate techniques such as Neural Network Potentials (NNPs), QM/MM methods, ab initio molecular dynamics, GPU computing, polarizable force fields, and enhanced conformational sampling methods.

Machine Learned Interatomic Potentials
Our group develops computational methods that predict the potential energy of chemical structures using machine learning methods like neural networks. These methods can provide the accuracy of the ab initio methods but at a computational cost that is millions of times lower. This makes it possible to simulate large, complex systems with unpresidented accuracy.

Intermolecular Interactions
Computer simulations require accurate representations of intermolecular interactions. In collaboration with the Johnson group at Dalhousie University, we are developing new representations of intermolecular interactions that describe dispersion interactions in matter more realistically. Recently, our group has begun to explore machine-learned neural network potentials as a radically different way to represent intermolecular interactions.
Mohebifar, M., Johnson, E.R., Rowley, C. N. J. Chem. Theory Comput., 2017, doi: 10.1021/acs.jctc.7b00522
S.-L. J. Lahey, C. N. Rowley, Simulating Protein-Ligand Binding with Neural Network Potentials. Chem. Sci., 2020, doi: 10.1039/C9SC06017K
Walters, E., Mohebifar, M., Johnson, E.R., Rowley, C. N., Evaluating the London Dispersion Coefficients of Protein Force Fields Using the Exchange-Hole Dipole Moment Model, J. Phys. Chem B. 2018, doi: 10.1021/acs.jpcb.8b02814

Covalent Modification of Proteins
Covalent-modifier drugs act on their target by forming a chemical bond with a side-chain of the targeted protein. These covalent modifiers account for 26% of enzyme-targeting drugs, including widely used drugs such as penicillin and aspirin. Recently, this mode of action has been used to develop a new class of anti-cancer drugs that contain an electrophilic group that forms a chemical bond with the target kinase.
Modelling the activity of these inhibitors requires a more sophisticated set of simulation tools than the tools that are used to model conventional reversible-binding drugs. These include pKa calculations to determine which amino acids are the most reactive and QM/MM simulations to model the chemical reaction between the drug and its target. We are currently studying the kinase family of proteins, which contain many important targets for anti-cancer drugs. Covalent-modifier drugs have the potential to improve the selectivity for target kinases.
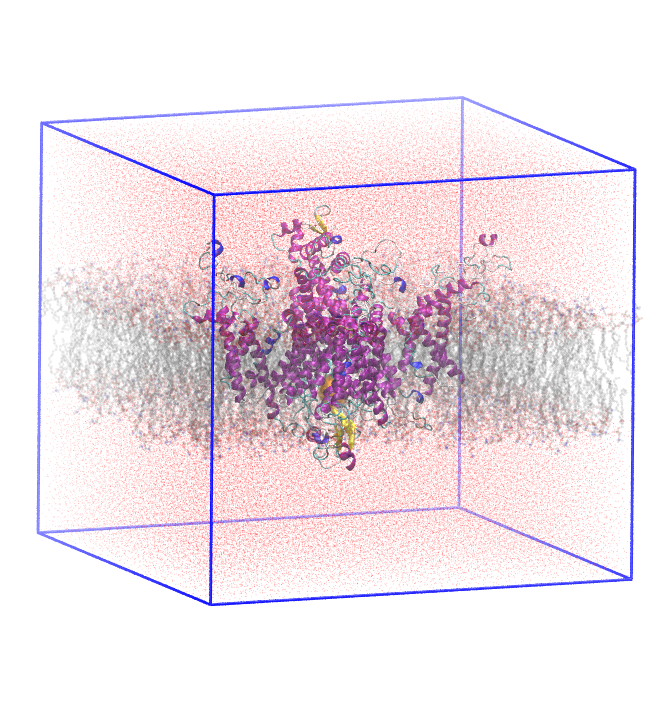
Simulations of NaV Channels
Our research uses molecular dynamics simulations to study small-molecule blockers of voltage-gated sodium (NaV) channels, which play a critical role in pain signalling. By modeling the interactions of these compounds with NaV channel isoforms at the atomic level, we aim to identify features that enhance selectivity and efficacy, guiding the design of novel therapeutics for chronic pain management.

Multiscale Simulations
The solvation of ions is central to biochemistry and marine chemistry. Our group has developed an interface between the molecular dynamics code CHARMM and the quantum chemistry program TURBOMOLE. This CHARMM-TURBOMOLE interface allows us to perform extended QM/MM molecular dynamics simulations using high-level QM methods and polarizable MM force fields. This work is performed in parallel to development of our polarizable force fields.
Riahi, S., Rowley C.N. The CHARMM-TURBOMOLE Interface for Efficient and Accurate QM/MM Molecular Dynamics, Free Energies, and Excited State Properties. J. Comput. Chem. 2014, 35, 2076–2086. doi: 10.1002/jcc.23716
Riahi, S., Roux, B., Rowley, C.N. QM/MM Molecular Dynamics Simulations of the Hydration of Mg(II) and Zn(II) Ions. Can. J. Chem. 2013, 91(7), 552–558